Що спричиняє розвиток хвороби Гоше?
Ферменти (ензими) — це біологічно активні речовини, що утворюються і використовуються організмом для нормального протікання хімічних реакцій.
При хворобі Гоше, один з ферментів ― глюкоцереброзидаза ― не утворюється у достатній кількості. Глюкоцереброзидаза робить можливим розщеплення жирової речовини під назвою глюкоцереброзид. За відсутності достатньої кількості чи активності ферменту, глюкоцереброзид накопичується всередині деяких клітин (макрофагів), в результаті чого останні збільшуються у розмірі. Ці великі клітини, що накопичують глюкоцереброзид, називаються «клітинами Гоше». Глюкоцереброзид накопичується у лізосомах клітин, через це хвороба Гоше відноситься до так званих лізосомних хвороб накопичення.
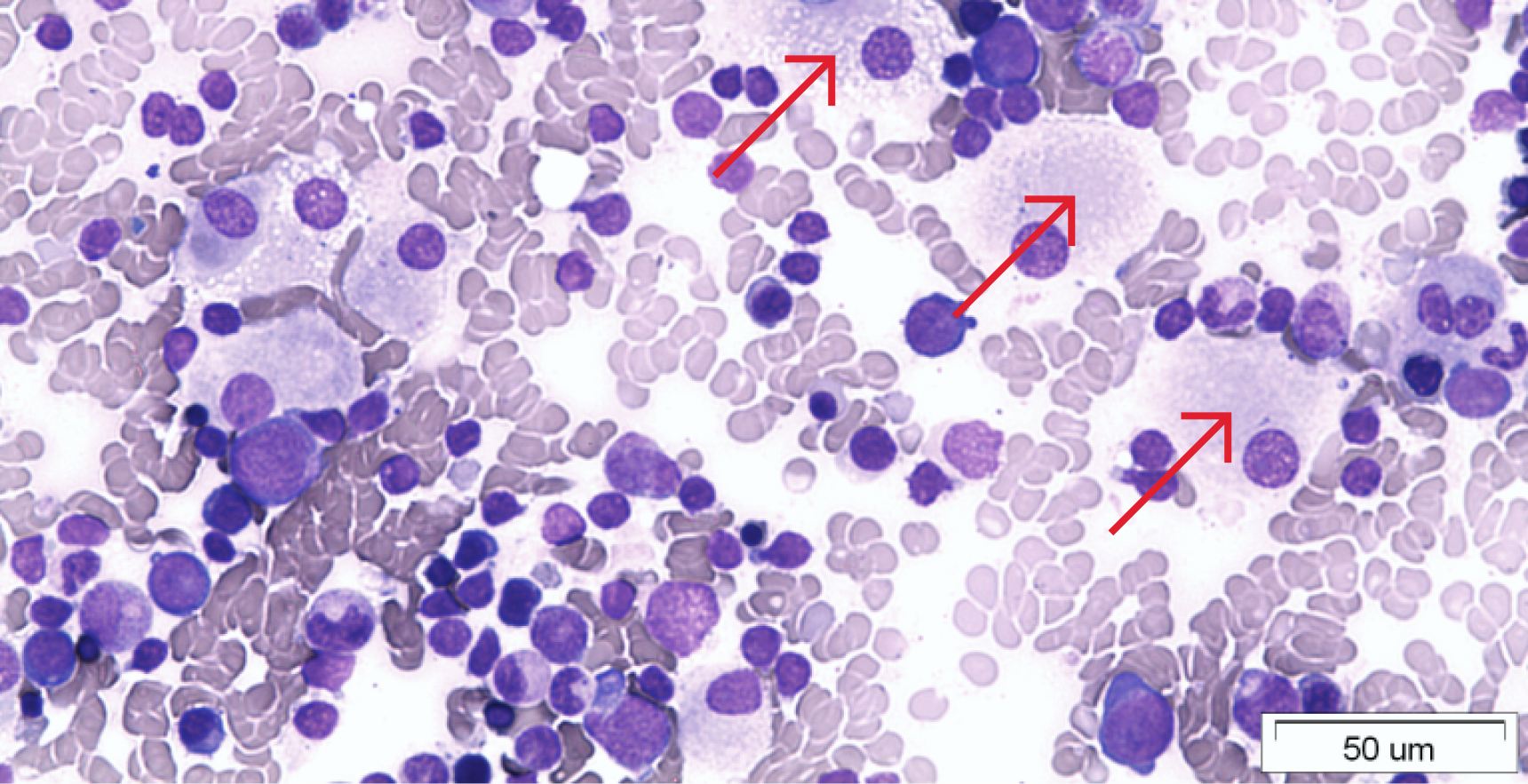
Клітини Гоше не здатні нормально функціонувати, і, по мірі того, як вони накопичуються, порушується функціонування відповідних органів та тканин.
Розуміння ключових слів
Успадкування хвороби Гоше
Як у пацієнтів виникає хвороба Гоше?
Гени ― це ділянки ДНК, на яких записана інформація про те, як клітинам слід виконувати свою роботу, іншими словами, це комп'ютерна програма для організму. Всі гени передаються нам від батьків. Те, чи є у людини хвороба Гоше, залежить від двох генів β-глюкоцереброзидази (або GBA: одного від батька, одного від матері.)
У пацієнтів з хворобою Гоше обидва гени GBA, які повідомляють клітинам про необхідність утворення фермента β-глюкоцереброзидази, містить мутацію, що означає, що в організмі виробляється менша кількість ферменту, або фермент, який виробляється, не функціонує належним чином. У деяких випадках, фермент взагалі не виробляється. Це призводить до накопичення жирової речовини (глюкоцереброзиду) у білих кров'яних клітинах (макрофагах), а отже і до проблем з функціонуванням різних органів тіла (див. розділи "Що таке хвороба Гоше" і "Що спричиняє розвиток хвороби Гоше").
Пацієнт з хворобою Гоше не може нічого вдіяти, щоб зупинити це захворювання, адже гени GBA, що він успадкував від батьків, не змінюються протягом життя.
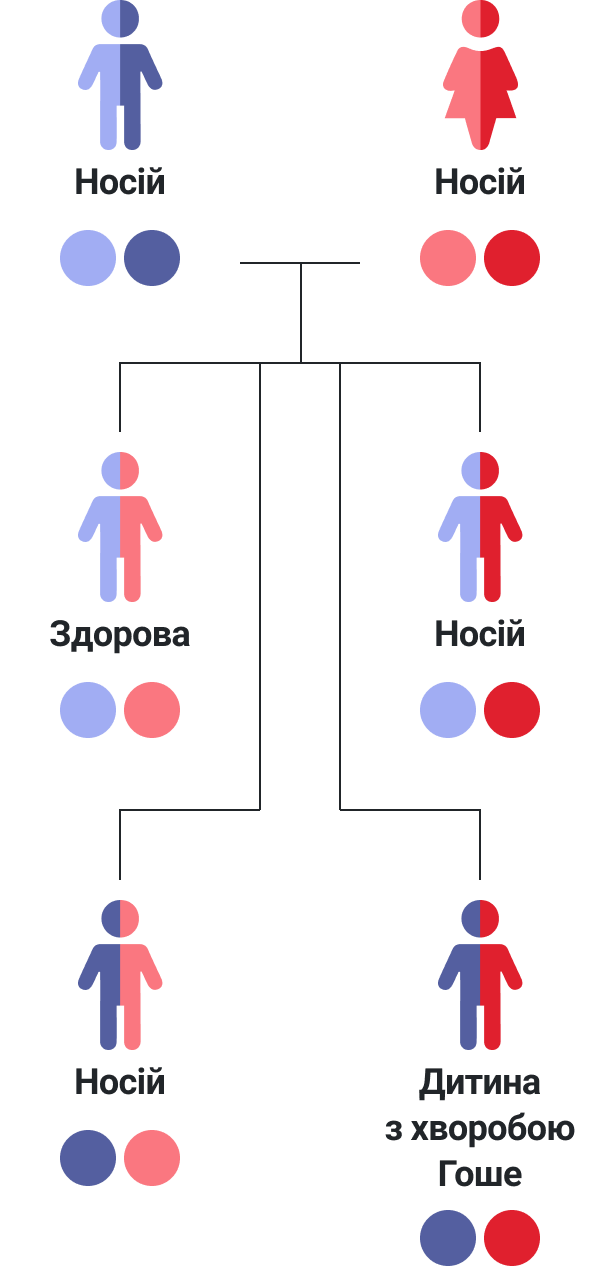
Яким чином хвороба Гоше передається в сім'ях?
У кожного з нас є два набори генів у всіх наших клітинах, – один набір ми отримуємо від матері, і один – від батька. Набір генів GBA, переданих (успадкованих) від батьків дитині, визначатиме те, чи матиме вона хворобу Гоше, чи ні:
— Якщо обидва гени в парі є нормальними, дитина не матиме хвороби Гоше
— Якщо обидва гени в парі є дефектними, у дитина виникне хвороба Гоше
— Якщо один ген є нормальним, а інший – дефектним, дитина не матиме хвороби Гоше, але буде її «носієм», тобто матиме ризик передати ген з мутацією наступним поколінням.
Для того, щоб у людини розвинулися ознаки і симптоми хвороби Гоше, вона повинна успадкувати дефектний ген GBA від обох своїх батьків, – це називається захворюванням з «аутосомно-рецесивним типом успадкування».
Носії не хворіють на хворобу Гоше самі, але вони мають один дефектний ген GBA і можуть передати його своїм дітям. Хоча носії хвороби Гоше мають один дефектний ген GBA, наявність у них іншого нормального гену GBA дозволяє організму утворювати фермент β-глюкоцереброзидазу у достатній кількості, через це шкідливого накопичення глюкоцереброзиду не відбувається. Попри те, що кількість фермента β-глюкоцереброзидази, що виробляється у них в організмі, як правило, нижче норми, носії не мають симптомів хвороби Гоше і можуть не знати про наявність у них одного дефектного гену GBA.
Ймовірність передачі хвороби Гоше від батьків до дітей
Хвороба Гоше успадковується за «аутосомно-рецесивним типом успадкування». Щоб зрозуміти, що це означає на практиці, розберемо приклад: уявіть, що чоловік і жінка обидва є носіями хвороби Гоше, – вони не хворіють на цю хворобу самі, але кожен з них має один нормальний, і один дефектний ген GBA. У цьому випадку кожного разу при народженні дитини:
— Існує імовірність 1 з 4 (тобто 25%) того, що дитина успадкує 2 нормальних гени GBA, і не матиме хвороби (і не буде носієм хвороби Гоше)
— Існує імовірність 2 з 4 (тобто 50%) того, що дитина успадкує 1 дефектний ген GBA і буде носієм хвороби Гоше (але не матиме хвороби)
— Існує імовірність 1 з 4 (тобто 25%) того, що дитина успадкує 2 дефектних гени GBA і у неї буде хвороба Гоше
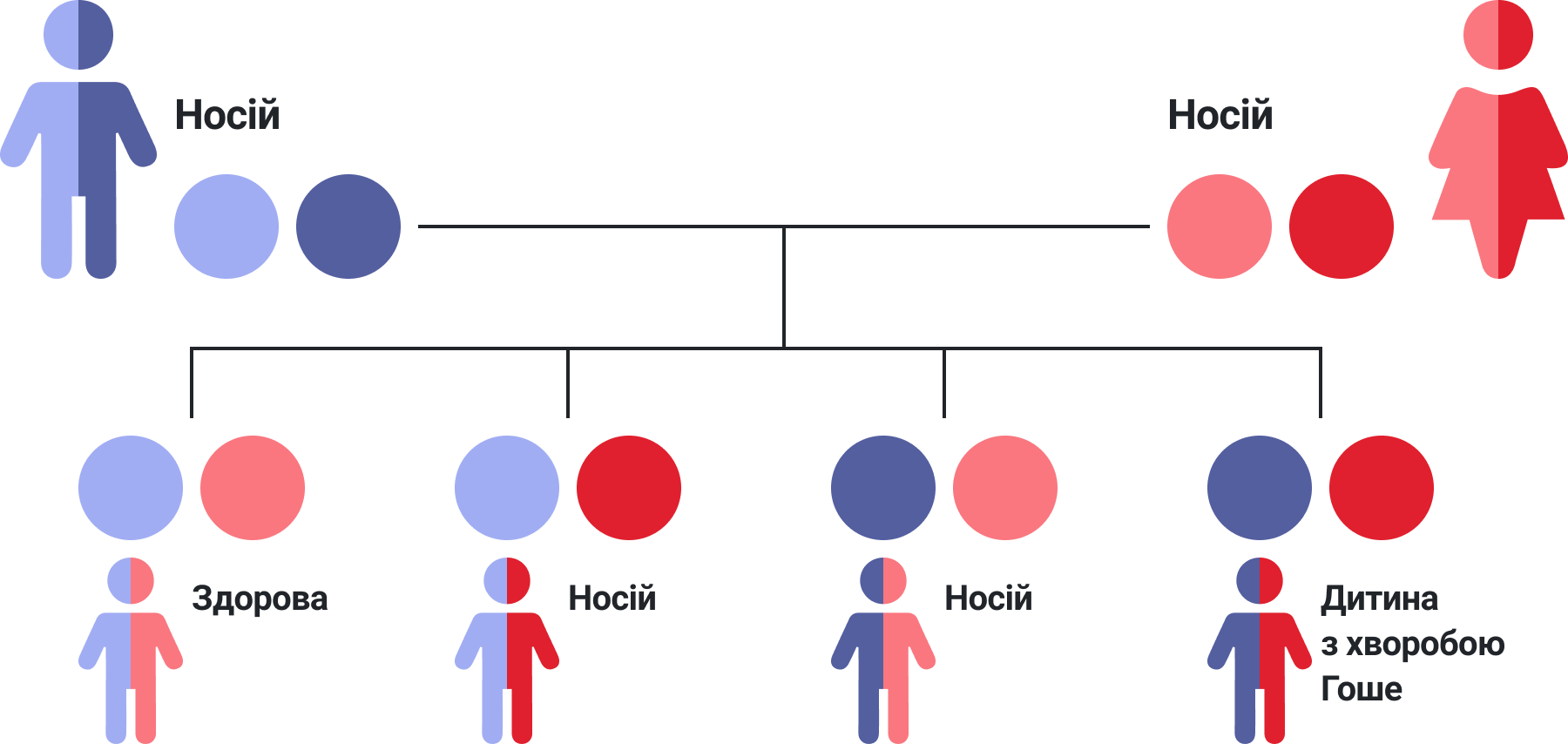
Якщо обидва батьки не хворіють на хворобу Гоше або не є носіями дефектного гена, з високою вірогідністю ніхто з їхніх дітей не буде уражений хворобою Гоше та не буде носієм.
Ознаки та симптоми
Прояви хвороби Гоше можуть відрізнятися у різних пацієнтів. У деяких пацієнтів перебіг захворювання є відносно легким, і захворювання може залишатися непоміченим до досягнення дорослого віку. У інших людей симптоми можуть розвиватися незабаром після народження.
Хворобу Гоше поділяють на три типи:
— Тип 1
- це найбільш поширена форма хвороби Гоше, яка не вражає нервові клітини пацієнта. Водночас, тип 1 може включати в себе ряд ознак і симптомів, таких як проблеми з кістками, зміни в аналізах крові, таким пацієнтам властива підвищена втомлюваність і збільшена селезінка. Хвороба також може вражати печінку і легені. Хворобу Гоше 1 типу називають "ненейропатичною -3"
— Тип 2
– це дуже рідкісна і тяжка форма хвороби, яка вражає лише немовлят. Симптоми з'являються через деякий час після народження і включають вже описані, але також до них додаються симптоми з боку нервової системи. Вони можуть включати косоокість, тугорухливість рук і ніг, судоми та утруднене ковтання. Цю форму хвороби називають «гострою нейропатичною хворобою Гоше»4. Хвороба Гоше 2 типу завжди призводить до летального наслідку, діти часто помирають до досягнення ними 2-річного віку. Це надзвичайно важка форма хвороби, яка на даний час не піддається лікуванню.
— Тип 3
Тип 3 також є рідкісною формою хвороби Гоше, що включає симптоми з боку нервової системи, але тип 3 не є настільки важким, як хвороба Гоше 2 типу. Перші симптоми можуть з’являтися як у немовлят, так і у дітей доросліше . Вони включають порушення рухливості очей і всі симптоми, описані вище. Хворобу цього типу називають «хронічною нейропатичною хворобою Гоше5».
Хоча поділ захворювання на три типи допоможе лікарям контролювати симптоми, перебіг хвороби Гоше значною мірою відрізняється у різних пацієнтів, і її часто розглядають як комплекси різних симптомів, від легких до важких, із збільшенням проблем з боку нервової системи6.
1. Cox TM, Schofield JP. Gaucher’s disease: clinical features and natural history. Bailleres Clin Haematol 1997; 10(4): 657-689.
2. Mistry PK, Sadan S, Yang R, Yee J, Yang M. Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am J Hematol 2007;82:697–701.
3. Damiano AM, Pastores GM, Ware JE Jr. The health-related quality of life of adults with Gaucher disease receiving enzyme replacement therapy: results from a retrospective study. Qual Life Res 1998;7(5):373-386.
4. Gupta N, Oppenheim I, Kauvar E, Tayebi N, Sidransky E. Type 2 Gaucher disease: phenotypic variation and genotypic heterogeneity. Blood Cells Mol Dis 2011;46(1):75-84.
5. Grabowski GA, Petsko GA, Kolodny EH. Chapter 146: Gaucher disease. Valle D, Beaudet AL, Vogelstein B, et al, eds. The Online Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw Hill; 2013. https://ommbid.mhmedical.com/content.aspx?bookid=2709&Sectionid=225546056. Last accessed August 20, 2024.
6. Sidransky E. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab 2004;83(1-2):6-15.
MAT-UA-2101034
23.08.2024