Болезнь
Фабри
Фабри
Что такое болезнь Фабри
Болезнь Фабри является редким наследственным прогрессирующим и мультисистемным заболеванием, сцепленным с Х-хромосомой, которое может поражать как мужчин, так и женщин любого возраста и этнического происхождения. Она была впервые описана в 1898 году независимо доктором Уильямом Андерсоном (Англия) и доктором Йоганнесом Фабри (Германия). Болезнь Фабри также известна как angiokeratoma corporis diffusum universal и болезнь Андерсона-Фабри1.
Патофизиологические механизмы,
лежащие в основе заболевания
лежащие в основе заболевания
Болезнь Фабри является одним из более 50 известных редких наследственных заболеваний, которые называются лизосомными болезнями накопления. Каждое из этих заболеваний обусловлено врожденным генетическим дефектом, приводящим к дефициту определенного лизосомного фермента или ферментов2. Возраст появления первых симптомов заболевания, поражения систем органов и степень тяжести этого заболевания в значительной степени варьируют, но все они являются прогрессирующими.
Болезнь Фабри вызвана мутацией в гене GLA, который кодирует лизосомальную α-галактозидазу A (также известную как α-GAL; α-Gal-A)1. Частичный или полный дефицит активности α-GAL приводит к пониженной способности катаболизировать липиды с терминальными α-галактозильными остатками. Эти липиды, особенно глоботриаозилцерамид (также известный как GL-3, Gb3), накапливаются в лизосомах многочисленных типов клеток в организме, включая эндотелиальные клетки капилляров, клетки почек, сердца и нервов, в результате вызывая прогрессирующее мультисистемное поражение. Со временем это может привести к циркуляторно-ишемическому поражению жизненно важных органов, таких как почки, сердце и/или цереброваскулярная система.
Накопление GL-3 в эндотелии капилляров
Стрелки указывают на участки накопления GL-3 на этом электронном микроснимке эндотелия капилляров почек у пациента с болезнью Фабри.
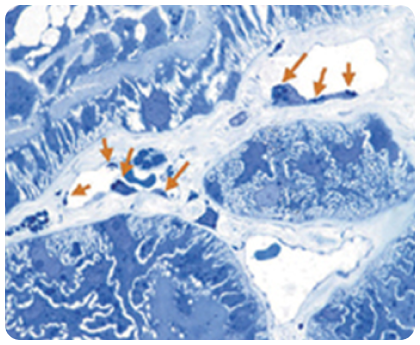
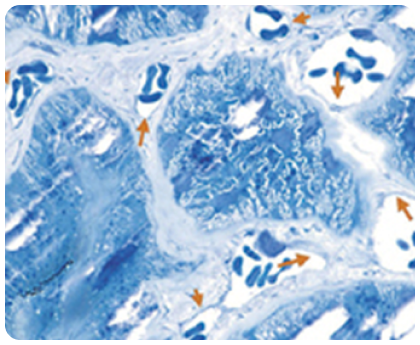
Изображение взято из галереи компании «Джензайм», используется с разрешения компании.
Манифестация болезни
Болезнь Фабри имеет широкий спектр гетерогенно прогрессирующих клинических фенотипов. Ранее был задокументирован только «классический» фенотип болезни Фабри. На сегодняшний день также сообщалось о неклассических фенотипах. В связи с таким широким спектром фенотипов клинические проявления могут в значительной степени варьировать в зависимости от разных пациентов. Клинические проявления болезни у пациентов могут включать нейропатическую боль, болевые кризы, непереносимость жары/холода, дерматологические проблемы (ангиокератомы), проявления со стороны органа зрения, расстройства со стороны желудочно-кишечного тракта, расстройства со стороны сердца (например, аритмии и гипертрофию левого желудочка (ГЛЖ)), проблемы с почками, инсульт. В связи с таким широким спектром клинических проявлений, диагностика болезни Фабри может быть сложной задачей.
Литература
1. Desnick R.J., Ioannou Y.A., Eng C.M. (2014). α-Galactosidase A Deficiency: Fabry Disease. In Valle D, Beaudet A.L., Vogelstein B, Kinzler K.W., Antonarakis S.E., Ballabio A, Gibson K, Mitchell G (Eds), . Retrieved December 11, 2015 from http://ommbid.mhmedical.com/content.aspx?bookid=971&Sectionid=62644837. Last accessed August 20, 2024.
2. Germain, DP. Fabry Disease. Orphanet J Rare Dis. 2010 Nov 22;5:30. doi: 10.1186/1750-1172-5-30.
MAT-UA-2101038
23.08.2024
23.08.2024