
Диагностика и подтверждающие исследования
Болезнь Помпе – это редкое заболевание с признаками и симптомами, которые могут имитировать многие другие заболевания.
Часто диагноз «болезнь Помпе» упускается из виду, по крайней мере, в первое время, до момента исключения других, более распространенных заболеваний. Результатом может стать опасная или даже опасная для жизни задержка диагностики. Ранняя диагностика имеет особое значение для малышей, поскольку отсутствие лечения в течение первого года жизни обычно приводит к смерти. Ретроспективный анализ малышей с болезнью Помпе обнаружил промежуток в 2,7 месяца между средним возрастом появления симптомов и моментом установления диагноза.13
Недавно проведенный анализ данных Реестра болезни Помпе обнаружил задержку диагностики у малышей, детей и подростков/взрослых. Среди младенцев, у которых симптомы появились в течение первых 12 месяцев жизни, а также была диагностирована кардиомиопатия (то есть классическая инфантильная болезнь Помпе), средняя задержка диагностики составила 1,4 месяца. Среди пациентов с началом заболевания в возрасте старше 12 лет, среднее время задержки диагностики составляло 6,0 лет. Самая длинная задержка диагностики составила в среднем 12,6 лет у пациентов, у которых симптомы появились в течение первых 12 месяцев жизни, но отсутствовала кардиомиопатия, или у которых симптомы появились в возрасте от 12 месяцев до 12 лет.14
Таким образом, существует необходимость как можно более ранней диагностики во всех возрастных группах пациентов с болезнью Помпе с целью оптимизации результатов лечения.
Задержка диагностики у новорожденных, детей и подростков/взрослых с болезнью Помпе14
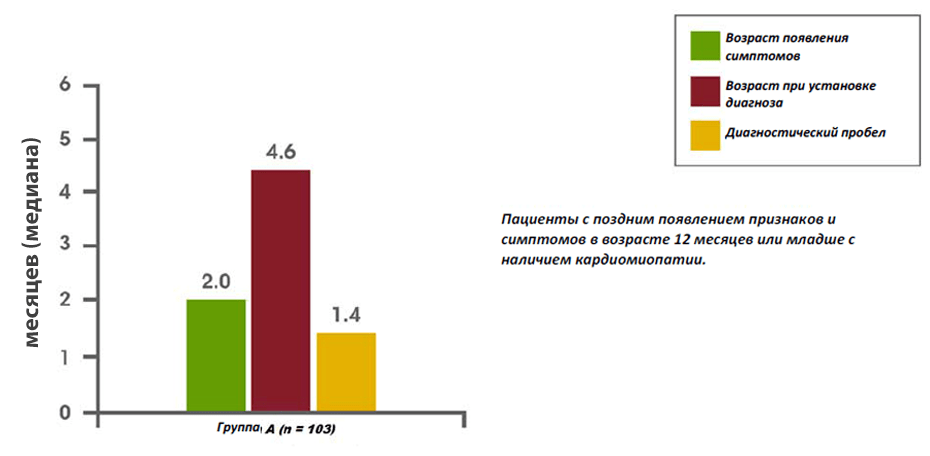
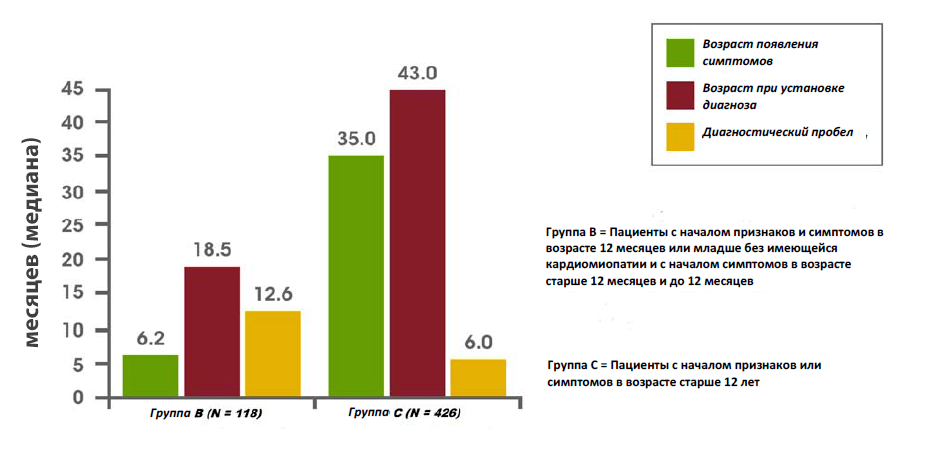
Взято из Kishnani et al, 201314
Алгоритм диагностики
Хотя клинические проявления диагностики болезни Помпе широко варьируются, обычно диагностический процесс включает:
- Клиническую оценку имеющихся симптомов семейным врачом
- Направление к узкому специалисту для дальнейшего клинического осмотра, в том числе - для проведения дополнительных лабораторных или клинических исследований
- Подтверждающее исследование
Окончательный диагноз
Болезнь Помпе подтверждается при полном отсутствии или выраженном снижении активности кислой альфа-глюкозидазы (GAA)15,16. В любом случае остаточная активность GAA у пациентов с болезнью Помпе может составлять от менее 1% (как правило, у малышей) до 30% от среднего уровня нормы.17 Болезнь Помпе также подтверждается с помощью генетического анализа с целью подтверждения наличия двух мутантных аллелей (см. раздел «Мутации»).
Традиционно определение уровня активности фермента GAA проводилось с применением культивированных фибробластов кожи.15,16 Тем не менее, взятие образцов является инвазивным, и результаты можно получить примерно через 6 недель. Такой длительный период ожидания является нежелательным, особенно для малышей, у которых заболевание слишком стремительно прогрессирует. Таким образом, значительное практическое значение приобретает использование образцов крови, в том числе - метода сухих капель крови. Исследование образцов крови с целью подтверждения диагноза болезни Помпе является минимально инвазивным, точным и позволяет получить результаты уже через несколько дней.15, 16 При обнаружении сниженной ферментной активности GAA, диагноз следует подтвердить с помощью исследования второго образца и/или методом генетического анализа гена GAA.5
Биопсия мышц является еще одним вариантом для исследования активности GAA, однако она нежелательна. Это связано с инвазивностью и высоким риском ложно-положительных результатов из-за неправильного обращения с образцом. Биопсия мышц может использоваться для гистологической оценки, однако следует отметить, что содержание гликогена может значительно варьироваться в разных мышцах, следовательно, на первый взгляд нормальные результаты при исследовании биоптата не исключают диагноз болезни Помпе.16 Таким образом, диагноз болезни Помпе всегда следует подтверждать или измерениями активности GAA, или с помощью генетического анализа.
Как исключить диагноз болезни Помпе у Вашего пациента
Обратитесь к представителю ООО "Фармакси" для получения карт для забора крови для проведения первичной лабораторной диагностики.
Список литературы
1. Kishnani PS, Steiner RD, Bali D, et al. Pompe disease diagnosis and management guideline. Genet Med 2006; 8:267-88.
2. Gilbert-Barness E. Review: Metabolic cardiomyopathy and conduction system defects in children. Ann Clin Lab Sci 2004; 34:15-34.
3. Howell RR, Byrne B, Darras BT, Kishnani P, Nicolino M, van der Ploeg A. Diagnostic challenges for Pompe disease: An under-recognized cause of floppy baby syndrome. Genet Med 2006:8;1-8.
4. Gilchrist JM. Overview of neuromuscular disorders affecting respiratory function. Semin Respir Crit Care Med 2002; 23:191-200.
5. American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM). Diagnostic criteria for late-onset (childhood and adult) Pompe disease. Muscle Nerve. 2009;40:149-60.
6. Ausems MG, Lochman P, van Diggelen OP, Ploos van Amstel HK, Reuser AJ, Wokke JH. A diagnostic protocol for adult-onset glycogen storage disease type II. Neurology. 1999 Mar 10;52(4):851-3.
7. Goldstein JL, Young SP, Changela M, Dickerson GH, Zhang H, Dai J, Peterson D, Millington DS, Kishnani PS, Bali DS (2009). Screening for Pompe disease using a rapid dried blood spot method: experience of a clinical diagnostic laboratory. Muscle Nerve 40:32-36.
8. Preisler N, Lukacs Z, Vinge L, Madsen KL, Husu E, Hansen RS, Duno M, Andersen H, Laub M, Vissing J (2013). Late-onset Pompe disease is prevalent in unclassified limb-girdle muscular dystrophies. Mol Genet Metab 110(3):287-9.
9. Willis T, Roberts M, Hilton-Jones D, Quinlivan R,5 Hanna M, Straub V (2012). Detection rate of Pompe disease in undiagnosed neuromuscular patients from four major centre’s in the UK- Results of a 12 month prospective audit. BMC Musculoskelet Disord 14(Suppl 2):P20.
10. Bautista Lorite J (2013) Detección de la enfermedad de Pompe en pacientes con distrofia de cinturas indefinidas o hiperCKemias asintomáticas. Expert Rev Neur Ed especial Octubre 2013:17-19. [Article in Spanish]
11. Fernandez C, de Paula AM, Figarella-Branger D, Krahn M, Giorgi R, Chabrol B, et al. (2006). Diagnostic evaluation of clinically normal subjects with chronic hyperCKemia. Neurology 66:1585-7.
12. Spada M, Porta F, Vercelli L, Pagliardini V, Chiadò-Piat L, Boffi P, Pagliardini S, Remiche G, Ronchi D, Comi G, Mongini T (2013). Screening for later-onset Pompe's disease in patients with paucisymptomatic hyperCKemia. Mol Genet Metab 109:171-173.
13. Kishnani PS, Hwu W-L, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr 2006; 148:671-676.
14. Kishnani PS, Amartino HM, Lindberg C, Miller TM, Wilson A, Keutzer J; Pompe Registry Boards of Advisors. Timing of diagnosis of patients with Pompe disease: data from the Pompe registry. Am J Med Genet A. 2013;161A(10):2431-43.
15. Zhang H, Kallwass H, Young SP, et al. Comparison of maltose and acarbose as inhibitors of maltase-glucoamylase activity in assaying acid alpha-glucosidase activity in dried blood spots for the diagnosis of infantile Pompe disease. Genet med 2006; 8:302-306.
16. Winchester B, Bali D, Bodamer OA, et al for The Pompe Disease Diagnostic Working Group. Methods for a prompt and reliable laboratory diagnosis of Pompe disease: report from an international consensus meeting. Mol Genet Metab. 2008;93(3):275-281.
17. van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet. 2008;372(9646):1342-53.