Хвороба
Фабрі
Фабрі
Що таке хвороба Фабрі
Хвороба Фабрі є рідкісним спадковим прогресуючим і мультисистемним захворюванням, зчепленим з Х-хромосомою, яке може вражати як чоловіків, так і жінок будь-якого віку та етнічного походження. Вона була вперше описана в 1898 році незалежно доктором Вільямом Андерсоном (Англія) і доктором Йоганнесом Фабрі (Німеччина). Хвороба Фабрі також відома як angiokeratoma corporis diffusum universal і хвороба Андерсона-Фабрі1.
Патофізіологічні механізми,
що лежать в основі захворювання
що лежать в основі захворювання
Хвороба Фабрі є однією з більш ніж 50 відомих рідкісних спадкових захворювань, які називаються лізосомними хворобами накопичення. Кожне з цих захворювань спричинене вродженим генетичним дефектом, що призводить до дефіциту певного лізосомного ферменту або ферментів2. Вік появи перших проявів захворювання, уражень систем органів і ступінь тяжкості цього захворювання значною мірою варіюють, але всі вони є прогресуючими.
Хвороба Фабрі спричинена мутацією в гені GLA, який кодує лізосомальну α-галактозидазу A (також відому як α-GAL; α-Gal-A)1. Частковий або повний дефіцит активності α-GAL призводить до зниженої здатності катаболізувати ліпіди з термінальними α-галактозильними залишками. Ці ліпіди, особливо глоботріаозилцерамід (також відомий як GL-3; Gb3), накопичуються в лізосомах численних типів клітин в організмі, включаючи ендотеліальні клітини капілярів, клітини нирок, серця і нервів, що в результаті викликає прогресуюче мультисистемне ураження. З часом це може призвести до циркуляторно-ішемічного ураження життєво-важливих органів, таких як нирки, серце та/ або цереброваскулярна система.
Накопичення GL-3 в ендотелії капілярів
Стрілки вказують на ділянки накопичення GL-3 на цьому електронному мікрознімку ендотелію капілярів нирок у пацієнта з хворобою Фабрі.
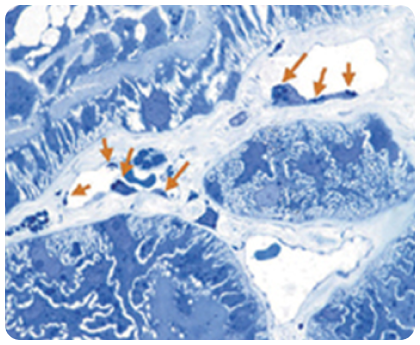
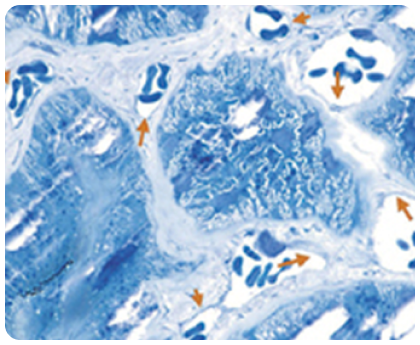
Зображення взято з галереї компанії «Джензайм», використовуэться з дозволу компанії.
Маніфестація хвороби
Хвороба Фабрі має широкий спектр гетерогенно прогресуючих клінічних фенотипів. Раніше було задокументовано лише «класичний» фенотип хвороби Фабрі. На сьогоднішній день також повідомлялося про некласичні фенотипи. В зв'язку з таким широким спектром фенотипів, клінічні прояви можуть значною мірою варіювати в залежності від різних пацієнтів. Клінічні прояви хвороби у пацієнтів можуть включати невропатичний біль, больові кризи, непереносимість спеки/ холоду, дерматологічні проблеми (ангіокератоми), прояви з боку органу зору, розлади з боку шлунково-кишкового тракту, розлади з боку серця (наприклад, аритмії та гіпертрофію лівого шлуночка (ГЛШ)) , проблеми з нирками, інсульт. В зв'язку з таким широким спектром клінічних проявів діагностика хвороби Фабрі може бути складним завданням.
Література
1. Desnick R.J., Ioannou Y.A., Eng C.M. (2014). α-Galactosidase A Deficiency: Fabry Disease. In Valle D, Beaudet A.L., Vogelstein B, Kinzler K.W., Antonarakis S.E., Ballabio A, Gibson K, Mitchell G (Eds), . Retrieved December 11, 2015 from http://ommbid.mhmedical.com/content.aspx?bookid=971&Sectionid=62644837. Last accessed August 20, 2024.
2. Germain, DP. Fabry Disease. Orphanet J Rare Dis. 2010 Nov 22;5:30. doi: 10.1186/1750-1172-5-30.
MAT-UA-2101038
23.08.2024
23.08.2024