
Діагностика та підтверджуючі дослідження
Хвороба Помпе — це рідкісне захворювання з ознаками та симптомами, які можуть імітувати багато інших захворювань.
Часто діагноз «хвороба Помпе» не береться до уваги, принаймні у перший час, до моменту виключення інших, більш поширених захворювань. Рання діагностика має особливе значення для малюків, оскільки відстність лікування протягом першого року життя зазвичай призводить до смерті. Ретроспективний аналіз малюків із хворобою Помпе виявив проміжок в 2,7 місяця між середнім віком появи симптомів і моментом встановлення діагнозу.13
Нещодавно проведений аналіз даних Реєстру хвороби Помпе виявив затримку діагностики у малюків, дітей та підлітків/дорослих. Серед немовлят, у яких симптоми з'явилися протягом перших 12 місяців життя, а також була діагностована кардіоміопатія (тобто класична інфантильна хвороба Помпе), середня затримка діагностики склала 1,4 місяці. Серед пацієнтів з початком захворювання у віці старше 12 років, середній час затримки діагностики становив 6,0 років. Найдовша затримка діагностики склала в середньому 12,6 років у пацієнтів, у яких симптоми з'явилися протягом перших 12 місяців життя, але була відсутня кардіоміопатія, або у яких симптоми з'явилися у віці від 12 місяців до 12 років.14
Таким чином, існує необхідність якомога ранньої діагностики у всіх вікових групах пацієнтів з хворобою Помпе з метою оптимізації результатів лікування.
Затримка діагностики у немовлят, дітей та підлітків/дорослих із хворобою Помпе14
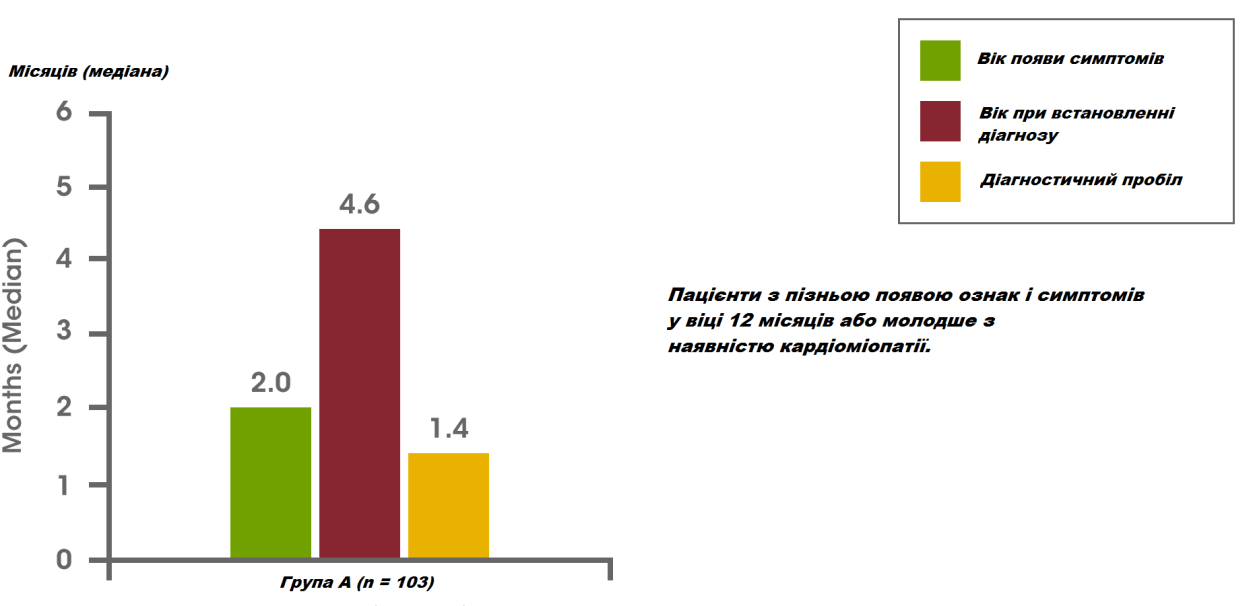
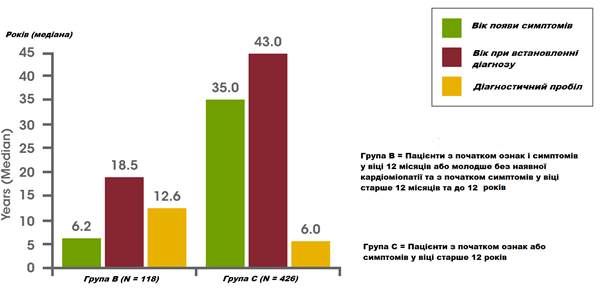
Взято з Kishnani et al, 201314
Алгоритм діагностики
Хоча клінічні прояви діагностики хвороби Помпе широко варіюють, зазвичай діагностичний процес включає:
- Клінічну оцінку наявних симптомів сімейним лікарем
- Направлення до вузького спеціаліста для подальшого клінічного огляду, у тому числі – для проведення додаткових лабораторних або клінічних досліджень
- Підтверджуюче дослідження
Остаточний діагноз
Хвороба Помпе підтверджується за повної відсутності або вираженого зниження активності кислої альфа-глюкозидази (GAA)15,16. У будь-якому випадку залишкова активність GAA у пацієнтів із хворобою Помпе може становити від менш ніж 1% (як правило, у малюків) до 30% від середнього рівня норми.17 Хвороба Помпе також підтверджується за допомогою генетичного аналізу з метою підтвердження наявності двох мутантних алелей (див. розділ «Мутації»).
Традиційно визначення рівня активності ферменту GAA проводилося із застосуванням культивованих фібробластів шкіри.15, 16 Тим не менш, взяття зразків є інвазивним, і результати можна отримати приблизно через 6 тижнів. Такий довгий період очікування є небажаним, особливо для малюків, у яких захворювання надто стрімко прогресує. Таким чином, значного практичного значення набуває використання зразків крові, у тому числі – методу сухих крапель крові. Дослідження зразків крові з метою підтвердження діагнозу хвороби Помпе є мінімально інвазивним, точним і дозволяє отримати результати вже через кілька днів.15, 16 При виявленні зниженої ферментної активності GAA, діагноз слід підтвердити за допомогою дослідження другого зразка та/або методом генетичного аналізу гена GAA.5
Біопсія м'язів є ще одним варіантом для дослідження активності GAA, однак вона є небажаною. Це пов'язано з інвазивністю та високим ризиком хибно-позитивних результатів через неправильне поводження зі зразком. Біопсія м'язів може використовуватися для гістологічної оцінки, проте слід зазначити, що вміст глікогену може значно варіювати у різних м'язах, отже, на перший погляд нормальні результати при дослідженні біоптату не виключають діагноз хвороби Помпе.16 Таким чином, діагноз хвороби Помпе завжди слід підтверджувати або вимірами активності GAA, або за допомогою генетичного аналізу.
Як виключити діагноз хвороби Помпе у Вашого пацієнта
Зверніться до представника ТОВ "Фармаксі" для одержання карт для забору крові для проведення первинної лабораторної діагностики.
Список літератури
1. Kishnani PS, Steiner RD, Bali D, et al. Pompe disease diagnosis and management guideline. Genet Med 2006; 8:267-88.
2. Gilbert-Barness E. Review: Metabolic cardiomyopathy and conduction system defects in children. Ann Clin Lab Sci 2004; 34:15-34.
3. Howell RR, Byrne B, Darras BT, Kishnani P, Nicolino M, van der Ploeg A. Diagnostic challenges for Pompe disease: An under-recognized cause of floppy baby syndrome. Genet Med 2006:8;1-8.
4. Gilchrist JM. Overview of neuromuscular disorders affecting respiratory function. Semin Respir Crit Care Med 2002; 23:191-200.
5. American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM). Diagnostic criteria for late-onset (childhood and adult) Pompe disease. Muscle Nerve. 2009;40:149-60.
6. Ausems MG, Lochman P, van Diggelen OP, Ploos van Amstel HK, Reuser AJ, Wokke JH. A diagnostic protocol for adult-onset glycogen storage disease type II. Neurology. 1999 Mar 10;52(4):851-3.
7. Goldstein JL, Young SP, Changela M, Dickerson GH, Zhang H, Dai J, Peterson D, Millington DS, Kishnani PS, Bali DS (2009). Screening for Pompe disease using a rapid dried blood spot method: experience of a clinical diagnostic laboratory. Muscle Nerve 40:32-36.
8. Preisler N, Lukacs Z, Vinge L, Madsen KL, Husu E, Hansen RS, Duno M, Andersen H, Laub M, Vissing J (2013). Late-onset Pompe disease is prevalent in unclassified limb-girdle muscular dystrophies. Mol Genet Metab 110(3):287-9.
9. Willis T, Roberts M, Hilton-Jones D, Quinlivan R,5 Hanna M, Straub V (2012). Detection rate of Pompe disease in undiagnosed neuromuscular patients from four major centre’s in the UK- Results of a 12 month prospective audit. BMC Musculoskelet Disord 14(Suppl 2):P20.
10. Bautista Lorite J (2013) Detección de la enfermedad de Pompe en pacientes con distrofia de cinturas indefinidas o hiperCKemias asintomáticas. Expert Rev Neur Ed especial Octubre 2013:17-19. [Article in Spanish]
11. Fernandez C, de Paula AM, Figarella-Branger D, Krahn M, Giorgi R, Chabrol B, et al. (2006). Diagnostic evaluation of clinically normal subjects with chronic hyperCKemia. Neurology 66:1585-7.
12. Spada M, Porta F, Vercelli L, Pagliardini V, Chiadò-Piat L, Boffi P, Pagliardini S, Remiche G, Ronchi D, Comi G, Mongini T (2013). Screening for later-onset Pompe's disease in patients with paucisymptomatic hyperCKemia. Mol Genet Metab 109:171-173.
13. Kishnani PS, Hwu W-L, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr 2006; 148:671-676.
14. Kishnani PS, Amartino HM, Lindberg C, Miller TM, Wilson A, Keutzer J; Pompe Registry Boards of Advisors. Timing of diagnosis of patients with Pompe disease: data from the Pompe registry. Am J Med Genet A. 2013;161A(10):2431-43.
15. Zhang H, Kallwass H, Young SP, et al. Comparison of maltose and acarbose as inhibitors of maltase-glucoamylase activity in assaying acid alpha-glucosidase activity in dried blood spots for the diagnosis of infantile Pompe disease. Genet med 2006; 8:302-306.
16. Winchester B, Bali D, Bodamer OA, et al for The Pompe Disease Diagnostic Working Group. Methods for a prompt and reliable laboratory diagnosis of Pompe disease: report from an international consensus meeting. Mol Genet Metab. 2008;93(3):275-281.
17. van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet. 2008;372(9646):1342-53.