
Патологія, що лежить в основі захворювання
Хвороба Помпе — це прогресуюче, мультисистемне, інвалідизуюче і потенційно смертельне нервово-м'язове захворювання.
Вперше ця хвороба була описана у 1932 році голландським патологом Іоганном К. Помпе, який оприлюднив випадок захворювання у 7-місячної дитини, що померла від ідіопатичної серцевої гіпертрофії1. У цієї дитини було виявлено масивне накопичення глікогену в багатьох тканинах організму, але переважно у скелетних і серцевих м'язах. У 1963 році був відкритий зв'язок захворювання зі спадковою недостатністю лізосомального ферменту, кислої альфа-глюкозидази (GAA)2, який відповідає за розщеплення глікогену до глюкози. Результатом такої недостатності є накопичення глікогену всередині лізосом, переважно у м'язових клітинах, що призводить до прогресуючої втрати м'язової функції.
Патогенез
Загальна інформація
Ген, розташований на хромосомі 17 (17q25.2-q25.3), кодує синтез кислої альфа-глюкозидази (GAA), ферменту, відповідального за розщеплення глікогену до глюкози всередині лізосом2,3. Мутації в цьому гені спричиняють виражений дефіцит або ж повну відсутність активності GAA, що призводить до накопичення глікогену всередині лізосом, переважно у м'язових клітинах3.
Пошкодження клітин та тканин
Постійне накопичення глікогену призводить до поступового збільшення у розмірах, набухання та розриву лізосом, що викликає ушкодження клітин. Це, в свою чергу, призводить до прогресуючої дегенерації скелетних і дихальних м'язів (разом із серцевим м'язом, насамперед у немовлят), що врешті-решт призводить до втрати їх функції2,4.
Наведене зображення, отримане при мікроскопічному дослідженні тканин, є прикладом накопичення глікогену та м'язової патології, що розвивається в результаті цього та часто виникає до появи будь-яких клінічно виявлених ознак або симптомів:
Знімки м'язової клітини новонародженої дитини, ураженої хворобою Помпе, отримані за допомогою електронного мікроскопа
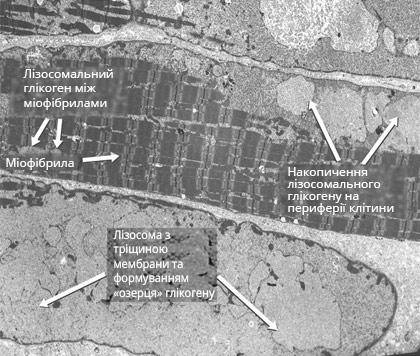
Це отриманий за допомогою електронного мікроскопа знімок скелетної м'язової клітини новонародженої дитини, картина характернаї для пацієнтів із хворобою Помпе. Накопичення глікогену призводить до збільшення та патологічного проникнення вмісту лізосом у клітинний простір. Хоча на початку розвитку захворювання можуть бути присутні деякі здорові міофібрили, з прогресуванням хвороби Помпе вони практично повністю замінюються глікогеном, що врешті-решт призводить до порушення м'язової функції. Взято з Thurberg et al., 20065.
Накопичення глікогену при хворобі Помпе, як правило, НЕ викликає аномалій метаболізму глюкози, таких як гіпоглікемія, оскільки глікоген, що зберігається у лізосомах, не бере участі у каскаді реакцій глюконеогенезу.
Одна патологія – різне прогресування захворювання
Варіабельність рівня ферментів
Оскільки хвороба Помпе завжди характеризується зниженням активності GAA, залишковий рівень ферментної активності відрізняється у різних пацієнтів і груп пацієнтів:
- У немовлят з хворобою Помпе зазвичай активність становить менш ніж 1% від середньостатистичної норми активності GAA6.
- У дітей та дорослих з хворобою Помпе може спостерігатися залишкова активність GAA на рівні 1−30% від середнього рівня норми6.
У цілому, існує слабка кореляція між залишковою активністю GAA та клінічними проявами хвороби у дітей та дорослих, хоча немовлята зазвичай мають більш важкі ураження.
Перебіг захворювання
У пацієнтів із класичною хворобою Помпе зазвичай спостерігається практично повна відсутність ферментної активності GAA, з вираженою кардіомегалією та швидким накопиченням глікогену в скелетних м'язах, що може в 10 разів перевищувати норму2. У цій групі пацієнтів захворювання прогресує дуже швидко і, як правило, за відсутності лікування призводить до летального кінця протягом першого року життя.7
У дітей та дорослих захворювання, як правило, прогресує з меншою швидкістю, ніж у класичних випадках у новонароджених пацієнтів, за незначного ураження серця або навіть без його ураження. Тим не менш, хвороба Помпе завжди невпинно прогресує та пов'язана зі значною захворюваністю та/або передчасною смертю8-10.
Список літератури
1. Pompe JC. Over idiopathische hypertrofie van het hart. Ned Tijdschr Geneeskd 1932; 76:304-311.
2. Hirschhorn R, Reuser AJ. Glycogen Storage Disease Type II: Acid α-Glucosidase (Acid Maltase) Deficiency. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G., eds. The Online Metabolic and Molecular Bases of Inherited Disease. OMMBID. Available at: http://ommbid.mhmedical.com/book.aspx?bookid=971 (link is external). Last accessed April 16, 2026.
3. Pittis MG, Filocamo M. Molecular genetics of late onset glycogen storage disease II in Italy. Acta Myol. 2007;26(1):67-71.
4. Muller-Felber W, Horvath R, Gempel K, Podskarbi T, Shin Y, Pongratz D, et al. Late onset Pompe disease: clinical and neurophysiological spectrum of 38 patients including long-term follow-up in 18 patients. Neuromuscul Disord. 2007.17;698-706.
5. Thurberg BL, Lynch Maloney C, Vaccaro C, Afonso K, Tsai AC, Bossen E, Kishnani PS, O'Callaghan M. Characterization of pre- and post-treatment pathology after enzyme replacement therapy for Pompe disease. Lab Invest. 2006;86(12):1208-20.
6. van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet. 2008;372(9646):1342-53.
7. Kishnani PS, Howell RR. Pompe disease in infants and children. J Pediatr 2004; 144(5 Suppl): S35-S43.
8. Hagemans ML, Winkel LP, Van Doorn PA, et al. Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients. Brain 2005; 128:671-7.
9. Wokke J, Escolar D, Pestronk A, Jaffe K, Carter G, van den Berg L, et al. Clinical features of late-onset Pompe disease: A prospective cohort study. Muscle Nerve 2008;38:1236-45.
10. Gungor D, de Vries JM, Hop WC, et al. Survival and associated factors in 268 adults with Pompe disease prior to treatment with enzyme replacement therapy. Orphanet J Rare Dis 2011; 6: 34