
Клінічні прояви
Хвороба Фабрі – мультисистемне захворювання з широким спектром гетерогенних клінічних фенотипів. Були описані класичні та некласичні (також відомі як атипові або з пізнім початком) варіанти хвороби Фабрі. При «класичній» хворобі Фабрі поява перших симптомів і клінічних ознак часто відмічається в дитинстві або підлітковому віці. На противагу цьому, при некласичній хворобі ознаки і симптоми з'являються пізніше протягом життя, хоча вони можуть значною мірою варіювати. В зв'язку з широким спектром клінічних фенотипів, задокументованих для обох цих форм захворювання, діагностика може бути достатньо складним завданням1,2.
У пацієнтів з хворобою Фабрі прогресуюче накопичення GL-3 починається на ранніх етапах життя і продовжується протягом десятиліть1,2. Ранній клінічний перебіг хвороби Фабрі зазвичай включає симптоми, які переважно впливають на якість життя пацієнтів, як показано на рисунку нижче. Прогресуюче накопичення GL-3 в лізосомах клітин організму може призвести до небезпечних для життя проявів, з ураженням нирок, серця, нервової системи1. Більш серйозні ускладнення з боку нирок, серця та цереброваскулярні захворювання, які можуть ставати небезпечними для життя, як правило, виникають до третього-п'ятого десятиліття2. В зв'язку з широким спектром і неспецифічним характером ознак і симптомів хвороби Фабрі, захворювання може залишатися нерозпізнаним, і/або такі ознаки і симптоми можна віднести на рахунок інших недуг. Це може призвести до пізньої діагностики хвороби, коли ураження життєво-важливих органів вже може бути наявним1. Діагностика на більш ранньому етапі може сприяти належному веденню пацієнтів з наявністю хвороби і контролю симптомів.
Аналіз даних перших 1765 пацієнтів, включених у Реєстр пацієнтів з хворобою Фабрі, показує, що типовий вік появи перших симптомів і діагностики хвороби становить 9 і 23 роки (особи чоловічої статі) і 13 і 32 роки (особи жіночої статі), відповідно, що вказує на затримки у встановленні діагнозу у осіб обох статей3.
Один або більше клінічних проявів з наведеного нижче списку можуть вказувати на наявність хвороби Фабрі. За їх наявності хворобу Фабрі необхідно включити в диференційний діагноз:
Ангіокератоми
Дисфункція серця
Симптоми з боку шлунково-кишкового тракту
Непереносимість спеки та холоду
Гіпогідроз/ ангідроз
Неврологічні прояви
Ураження органу зору
Біль
Прояви з боку нирок
Хвороба Фабрі також асоціюється з низкою більш загальних ознак і симптомів, включаючи погіршення добробуту і якості життя, зниження шкільної успішності/ продуктивності праці та здатності займатися іншими видами діяльності на дозвіллі; психосоціальний та поведінковий дефіцити; і незадовільний набір маси тіла2.
Рисунок 1. Ускладнення хвороби Фабрі
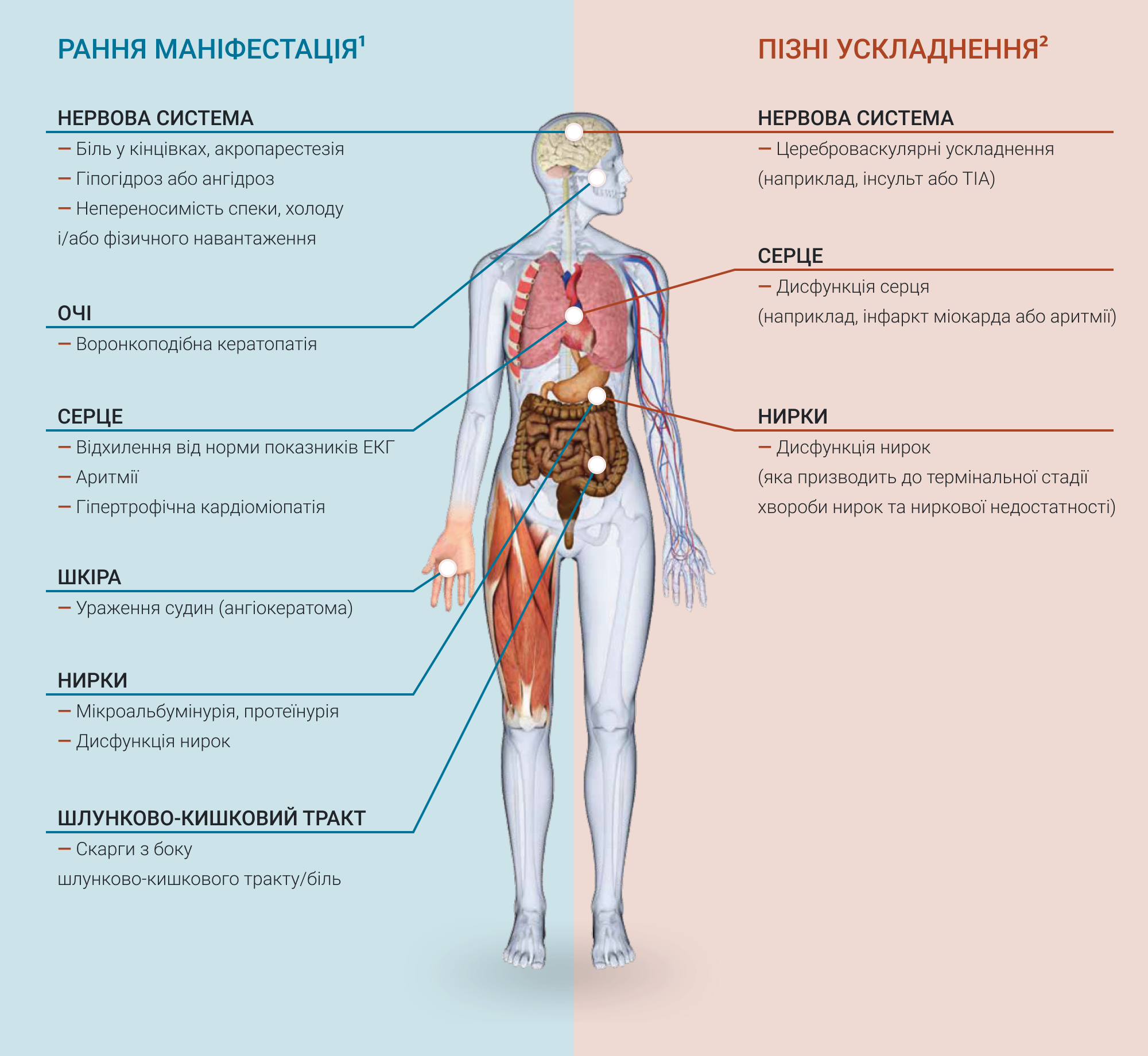
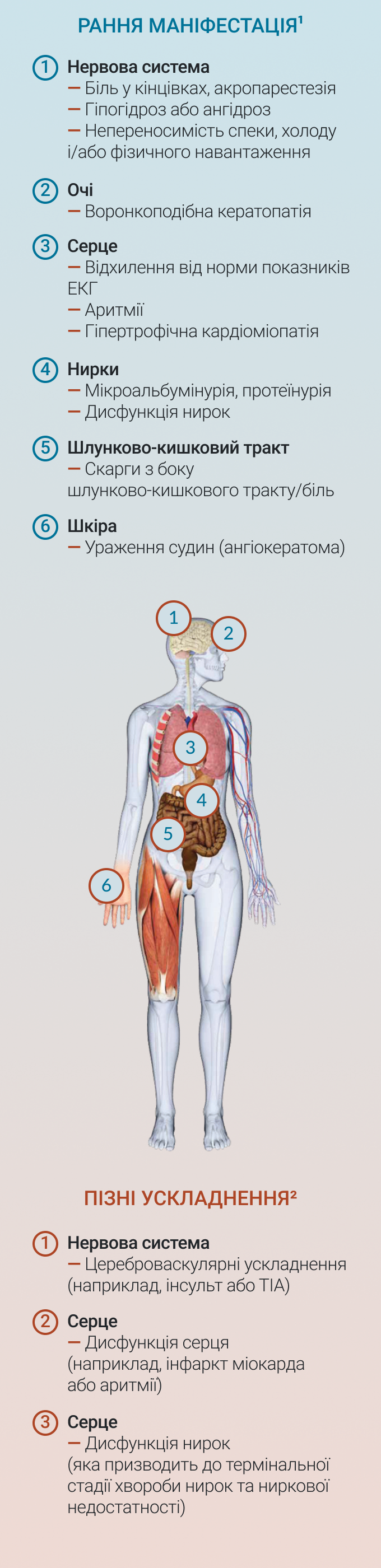
* Хвороба Фабрі також асоціюється з низкою більш загальних ознак і симптомів, таких як погіршення добробуту і якості життя; зниження шкільної успішності/ продуктивності праці; зниження здатності займатися іншими видами діяльності на дозвіллі, включаючи спорт; психосоціальний та поведінковий дефіцити; незадовільний набір маси тіла.
Прогресування та прогноз захворювання
Прогресуючі ураження органів та тканин, що асоціюються з хворобою Фабрі, можуть призвести до суттєвого зниження очікуваної тривалості життя. Як показано на рисунку нижче, медіана кумулятивної виживаності була зареєстрована як 50 років у чоловіків і 70 років у жінок, що представляє 20-річне та 15-річне зменшення (відповідно) тривалості життя порівняно з такою серед населення загалом3,4.
Виживаність:
пацієнти чоловічої статі з хворобою Фабрі Медіана виживаності у пацієнтів чоловічої статі з хворобою Фабрі становить 50 років, що відповідає скороченню тривалості життя на 20 років у порівнянні із загальною популяцією.
пацієнти чоловічої статі з хворобою Фабрі Медіана виживаності у пацієнтів чоловічої статі з хворобою Фабрі становить 50 років, що відповідає скороченню тривалості життя на 20 років у порівнянні із загальною популяцією.
Виживаність:
пацієнти жіночої статі з хворобою Фабрі Медіана виживаності у пацієнтів жіночої статі з хворобою Фабрі становить 70 років, що відповідає скороченню тривалості життя на 15 років у порівнянні із загальною популяцією.
пацієнти жіночої статі з хворобою Фабрі Медіана виживаності у пацієнтів жіночої статі з хворобою Фабрі становить 70 років, що відповідає скороченню тривалості життя на 15 років у порівнянні із загальною популяцією.
Оцінка впливу хвороби Фабрі на середню тривалість життя з використанням
методу оцінки виживаності Каплана-Мейєра.
методу оцінки виживаності Каплана-Мейєра.
Адаптовано за матеріалами MacDermot KD, et al. J Med Genet 2001;38(11):750–60 and MacDermot KD, et al. J Med Genet 2001;38(11):769–75.
Література
1. Desnick R.J., Ioannou Y.A., Eng C.M. (2014). α-Galactosidase A Deficiency: Fabry Disease. In Valle D, Beaudet A.L., Vogelstein B, Kinzler K.W., Antonarakis S.E., Ballabio A, Gibson K, Mitchell G (Eds), . Retrieved December 11, 2015 from http://ommbid.mhmedical.com/content.aspx?bookid=971&Sectionid=62644837. Last accessed August 20, 2024.
2. Germain DP. Fabry Disease. Orphanet J Rare Dis. 2010;5:30.
3. MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet 2001;38:769-75.
4. MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet 2001;38:750-60.
5. Wanner, C. Fabry disease model: a rational approach to the management of Fabry disease.Clin Ther. 2007;29 Suppl A:S2-5.
MAT-UA-2101038
23.08.2024
23.08.2024