Гоше
Загальний опис захворювання
Патологія, що лежить в основі захворювання
Хвороба Гоше — це лізосомальна хвороба накопичення, яка спричинена мутацією у гені ферменту β-глюкоцереброзидази (кислої β-глюкозидази). Наразі вже ідентифіковано більше 300 мутантних алелів, що асоційовані з цією хворобою.1
Хвороба вперше була описана в 1882 році французьким студентом-медиком Філіпом Гоше (Phillipe Gaucher) у молодої жінки, яка мала збільшену селезінку. Він помітив, що у тканині селезінки цієї пацієнтки зустрічаються великі нетипові клітини з накопиченням субстрату всередині.2 Більше 50 років потому французська вчена-хімік Генрієтта Агіон (Henriette Aghion) з’ясувала, що цим субстратом, що викликає збільшення селезінки та печінки, є різновід сфінголіпідів — глюкоцереброзид (глюкозилцерамід). І лише у 1965 році Роско Бреді (Roscoe Brady) з колегами довів, що хвороба Гоше спричинена зниженням активності глюкоцереброзидази.
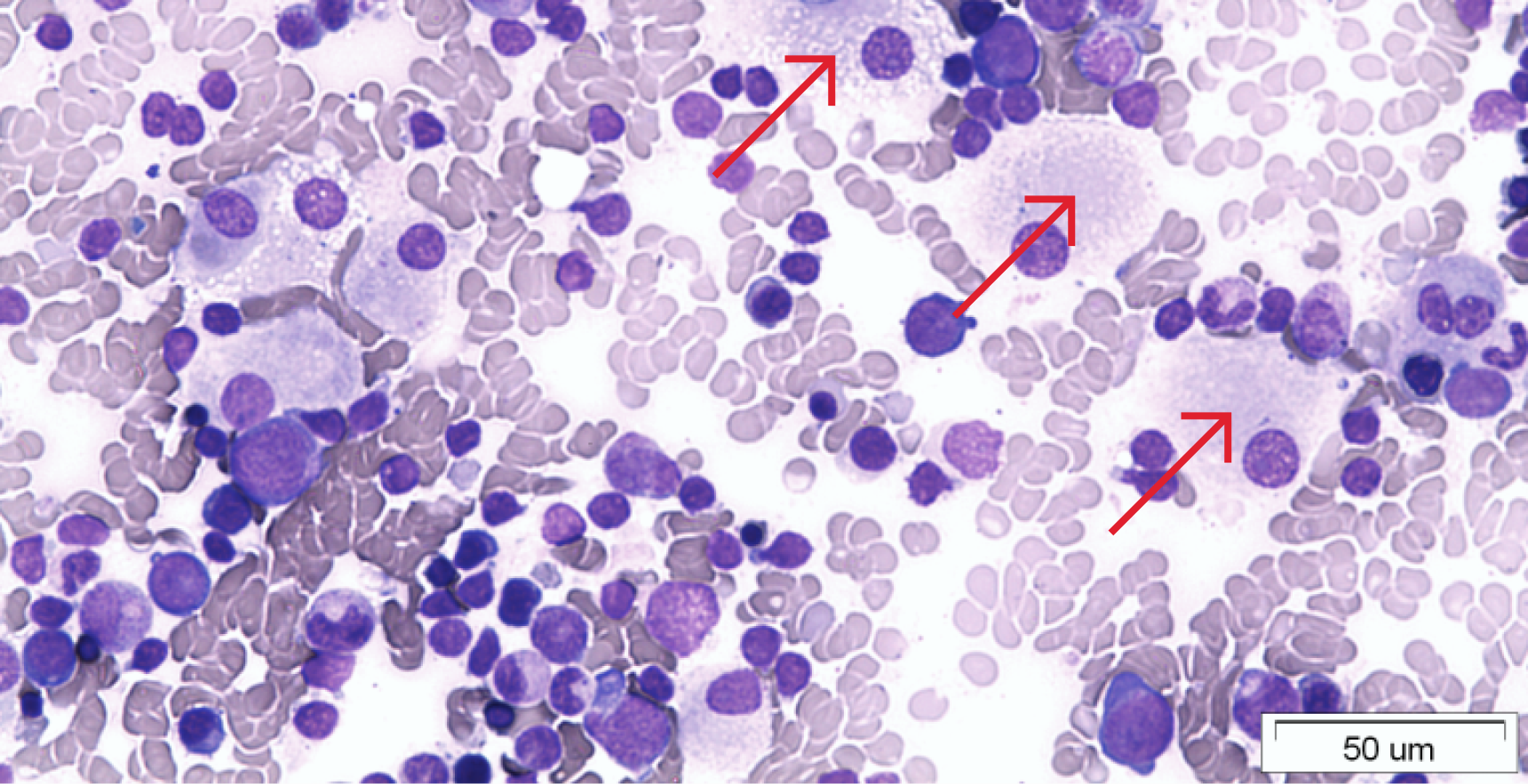
У нормі β-глюкоцереброзидаза розщеплює глікосфінголіпіди (а саме — глюкоцереброзид), що утворюються у результаті метаболізму структурних молекул клітинних мембран, зокрема мембран клітин крові. Однак через генетичну мутацію активність β-глюкоцереброзидази знижується, і глюкоцереброзид починає накопичуватися у лізосомах клітин, переважно в макрофагах. Макрофаги з істотним накопиченням глюкоцереброзиду — клітини Гоше — акумулюються в органах тіла (див. рис. 1). Утворення та скупчення клітин Гоше запускають подальший каскад патофізіологічних явищ, у тому числі призводять до хронічного запалення і виникнення гіперметаболічного стану.3
Хвороба Гоше ― мультисистемне захворювання, що характеризується значною варіабельністю клінічних проявів, ступеня важкості та перебігу. Частковий дефіцит β-глюкоцереброзидази супроводжується розвитком захворювань печінки, селезінки, кісткового мозку і легенів. Значний дефіцит ферменту також супроводжується і неврологічними проявами.4
1. Mistry PK, Weinthal JA, Weinreb NJ. Disease state awareness in Gaucher disease: a Q&A expert roundtable discussion. Clin Adv Hematol Oncol. 2012; 10:1-16.
2. Brady RO. Gaucher's disease: past, present and future. Bailleres Clin Haematol 1997;10(4):621-34.
3. Grabowski G. Gaucher disease and other storage disorders. Hematology Am Soc Hematol Educ Program 2012;1:13-18 (doi: 10.1182/asheducation-2012.1.13)
4. Cox TM, Schofield JP. Gaucher's disease: clinical features and natural history. Bailleres Clin Haematol 1997; 10(4) :657-689.
MAT-UA-2101034
23.08.2024